Molecular Genome Analysis
- Functional and Structural Genomics

Prof. Dr. Stefan Wiemann
Head of Division
Cancer and many other human diseases arise from genetic aberrations that are either inherited or occur spontaneously in somatic cells. These defects cause abnormal activities of gene products and lead to malfunctioning of molecular and cellular interactions which may induce tumors and cause cancer progression.
Image: Breast epithelial cells growing in a 3D matrix. Nuclei (blue), cytoskeleton (green) and Golgi (red) are stained. While the cells at the rim of the structure are polarized, the cells in the center are not. © dkfz.de,
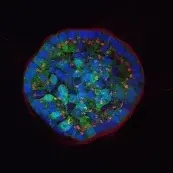
Image: Breast epithelial cells growing in a 3D matrix. Nuclei (blue), cytoskeleton (green) and Golgi (red) are stained. While the cells at the rim of the structure are polarized, the cells in the center are not. © dkfz.de,
Our Research
The central objective of our division is to understand the complexity of molecular mechanisms in the regulation of signaling networks and how these impact cancer development, metastasis, and drug resistance. To this end, we generate and maintain resources for large-scale experimentation, apply high-throughput functional genomics and proteomics technologies, and analyze candidate genes using in vitro as well as in vivo systems.
Effects of perturbations (gene gain- and loss-of-function, miRNA, drugs) imposed on the signaling processes are experimentally tested and then computationally modeled. This generates mechanistic knowledge that is exploited to identify new diagnostic and prognostic markers as well as to develop novel strategies for therapeutic intervention.
Our major focus here is on breast cancer, where we investigate protein and non-protein factors that are involved in the progression of different subtypes via their activities in interrelated signaling networks, and in the context of cell types within the tumor microenvironment.
Projects

Altered autocrine and paracrine signaling controls drug effects
Intra- and inter tumor heterogeneity are key factors affecting drug efficacy in individual patients. Mechanisms helping tumor cells persist drug treatment require immediate adaptation while long-term drug exposure establishes and fixes resistance states. We research on both, short-time drug effects and on long time resistance development, and there uncover molecular mechanisms underlying tumor cell survival.
Understanding failure of neoadjuvant chemotherapy in TNBC
The tumor microenvironment (TME) or tumor stroma comprises all cell types and extracellular matrix (ECM) that surrounds the tumor cells, jointly forming the tumor mass. The stromal compartment is comprised of immune cells from both, the innate and the adaptive systems, vascular cells, mesenchymal stem cells (MSC), cancer associated fibroblasts (CAF), and several other cell types. The TME affects tumor aggressiveness and the way tumor cells respond to therapies.
We have shown that primary patient-derived cancer associated fibroblasts (Berdiel-Acer et al. 2021 Oncogene, 40:2651-66) support tumor cell recovery from the impact of clinically applied chemotherapeutic drugs in breast cancer model systems (Maia et al. 2021 Mol Oncol, 15:1308-29). This process involves interferon beta signaling, an antiviral response, and expression of interferon stimulated genes.
Expression of the ISG protein OAS1 significantly correlated with residual disease (i.e., non-pCR) in the TNBC subtype of breast cancer after neoadjuvant chemotherapy (NACT), indicating clinical relevance of our findings (Bauer et al. 2022 Cancer Res, 82:P1-08-15).
miRNAs and isomiRs are non-coding determinants of cancer biology
The division has a long history for making discoveries in the field of miRNA-tumor interactions (e.g., Uhlmann et al., 2010 Oncogene, 30:4297-306, Uhlmann et al., 2012 Mol Syst Biol, 8:570, Körner et al., 2013 JBC, 288:8750-61; Keklikoglou et al., 2015 Oncogene, 34:4867-78; Breunig et al., 2018 Mol Oncol, 12(8):1447-63).
More recently, we determined previously unknown roles of miRNAs using a targeted proteomic screening approach and identified miR-193b to coordinately regulate Wnt/b-catenin, c-Met, and integrin signaling in aggressive triple-negative breast cancer (Giacomelli et al, 2021, BMC Cancer, 21(1):1296).
Expanding the scope of our studies also to the role of 5'isomiRs, we uncovered a negative feedback loop between a specific 5'isomiR of miR-183-5p and E2F1 (Li et al, 2022, J Exp Clin Cancer Res, 41(1):190). Clinical relevance of these findings was established with the help of data from The Cancer Genome Atlas (TCGA). Along these lines, a batch-correction strategy for reliable analysis of that data was developed (Ibing et al, 2021, NAR Cancer, 3(1):zcab007). Within a DFG-funded project, we currently investigate the mechanisms and the functional relevance of aberrant 5'isomiR processing and aberrant miRNA arm usage in cancer. Here, we could demonstrate that miR-1307-5p specifically counteracts the oncogenic functions of miR-1307-3p by inhibiting angiogenesis (Sumer OE et al, 2025, BMC Biol, 23:25).
Mechanisms of endocrine therapy resistance
Using in vitro models, we induced resistance to the ER-modulator tamoxifen as well as to long-term estrogen deprivation to mimic clinical aromatase inhibition, which are applied in pre- and postmenopausal patients, respectively. These models have since been utilized aimed at characterizing driver mechanisms of resistance (Borgoni et al. 2020 Cancers, 12(10):2918) and to understand the involvement of epigenetic mechanisms (Soleimani Dodaran et al. 2020 BMC Cancer, 20:676).
Using cellular barcoding and omics technologies, we uncovered alterations in resistant cell clones pointing at inter- (between cell lines) and intra-tumor (between different clones from the same cell line) heterogeneity (Beumers et al. 2023 NPJ Breast Cancer 9(1):97). We currently work on a candidate gene that might connect endocrine resistance with metabolic and epigenetic changes.
Modulations of EGFR-signaling via growth factors and therapeutic drugs
Receptor tryrosine kinase signaling via the EGF-receptor family of RTKs is a central signaling path also in breast cancer. Amplification of receptors (ERBB2) and mutations in signaling (like PIK3CA, RAS/RAF) are key events in different subtypes. We use targeted therapeutics (i.e., inhibitors and therapeutic antibodies) to better understand the wiring and rewiring in disease conditions. For relative quantification of protein activation states, we employed a targeted proteomics approach using Reverse Phase Protein Array (RPPA) technology (e.g., Sonntag et al. 2014 Transl Prot, 2:52-9, Bernhardt et al. 2017 Breast Cancer Res, 19(1):112, Bernhardt et al. 2019 J Prot Res, 18(3):1352-62; Byron et al. 2020 Sci Rep, 10(1):21985).
With funding by the BMBF (e:Med) we performed time-course analysis of cellular responses to combinations of different activators and inhibitors of the EGFR signaling network and developed a universal mathematical model that can predict the effects growth factors and inhibitors have in breast cancer subtypes.
In another time-course study, we found that glutamate ammonia ligase (GLUL) expression was negatively affected by hypoxia, and that this was associated with aggressive phenotypes in breast cancer in vitro, in vivo, and in patients (Bernhardt et al. 2019 J Proteome Res, 18(3):1352-62).
Collaborations
Signaling: Yosef Yarden (Rehovot), Moshe Oren & Yael Aylon (Rehovot), Pernette Verschure (Amsterdam), Luca Magnani (London), Niels de Jonge, Diana Peckys (Saarbrücken)
Breast cancer organoids: André Koch (Tübingen)
Patient Samples: PATH Biobank
Autophagy: Silvia Vega Rubin de Celis (Essen)
Bioinformatics: Lars Feuerbach, Benedikt Brors (DKFZ), Tim Beissbarth (Göttingen)
Mathematical modeling: Jens Timmer (Freiburg)
Clinics: Martina Vetter, Eva Kantelhardt, Christoph Thomssen (Halle/Saale)
Proteomics: Dominic Helm (DKFZ)

Coding and non-coding genomic drivers
Cancer is commonly regarded as 'a disease of the genes'. Alterations in abundance (copy number variation – CNV) or sequence (single nucleotide variation – SNV, insertions, deletions) and larger genomic rearrangements indeed affect individual or many genes thus contributing to disease onset and progression.
Non-coding genomic cancer drivers
While the focus has always been on coding cancer driver mutations, we believe that also non-coding SNVs have the potential to influence a cancer's phenotype. One prominent example are two highly recurrent SNVs in the promoter of the TERT gene which lead to its re-expression in cancer cells. Beyond that, only a few recurrent and functionally relevant promoter SNVs have been described. Therefore, we specifically focus on rare promoter SNVs. Within a collaborative project with the division of Applied Bioinformatics at DKFZ (Lars Feuerbach and Benedikt Brors) funded by the Fritz-Thyssen-Stiftung, we have identified and validated a number of rare functional promoter mutations activating downstream gene expression to potentially drive cancer progression.
Genomic drivers of rare cancer entities
With Florian Haller and Arndt Hartmann (University Clinics Erlangen) we have unveiled and experimentally verified molecular driver alterations in rare tumor entities. Enhancer hijacking activates gene expression of transcription factor NR4A3 as a driver mechanism in acinic cell carcinoma of the salivary gland (Haller et al. 2019 Nat Commun, 10(1):368; Haller et al. 2019 Am J Surg Pathol, 43(9):1264-72; Haller et al. 2020 Am J Surg Pathol, 44(9):1290-92).
We discovered NAB2-STAT6 fusion genes as drivers in lipomatous solitary fibrous tumors (Barthelmess et al., 2014 Am J Pathol, 184(4):1209-18). More recently, we went further into the molecular mechanisms associated this fusion event (Bieg et al. 2021 Am J Pathol, 191(4):602-17; Haller et al. 2021 Am J Pathol, 191(7):1314-24). Within a large sequencing study, carried out with the Charité and University Clinics Ulm, we discovered specific genomic as well as transcriptional alterations that drive primary CNS lymphoma and distinguish this entity from other B-cell lymphomas (Radke et al. 2022, Nat Commun, 13:2558).
Genomic drivers in KRAS- and BRAF-mutant MSS vs. MSI colorectal carcinoma
DKFZ has engaged with the Athens Comprehensive Cancer Center (Alex Pintzas, Olga Papadodima of the National Hellenic Research Foundation; Georgios Zografos of the Gennimatas General Hospital) in the frame of a Helmholtz European Partnering. Within this collaboration, we focus on the biology of colorectal cancer, where the microsatellite status (MSI vs. MSS) is a major determinant for therapy decision. We performed WES and RNA-seq with tumors from Greek CRC patients and did integrative analysis also in combination with TCGA and CPTAC CRC data, revealing the interplay of MSI/MSS with KRAS/BRAF mutation status and of affected signaling pathways as well as transcription factors. In this study, alterations were classified using a computational score for integrative cancer variant annotation and prioritization, while pathway and transcription factor activities were estimated in the context of the transcriptional and mutational circuits to identify potential vulnerabilities in individual tumors that might have implications for diagnosis and treatment (Vlachavas et al., 2025 Mol Oncol).
While our focus is mostly on colorectal carcinoma in this collaboration, we have collaborated with ACCC groups also in related projects (Mitra et al. 2021 Int J Cancer, 148(8):1993-2009; Berdiel-Acer et al. 2021 Oncogene, 40(15):2651-2666; Koralli et al. 2021 Mater Chem Front, 5(13):4950-4962).
Collaborations
Sarcoma/carcinoma: Florian Haller, Abbas Agaimy, Arndt Hartmann (Erlangen)
Breast cancer: Martina Vetter, Eva Kantelhardt, Christoph Thomssen (Halle/Saale), Christel Herold-Mende, Andreas Schneeweiss, Clarissa Gerhäuser (Heidelberg), Jens Timmer (Freiburg), Tim Beissbarth (Göttingen)
Primary CNS Lymphoma: Christel Herold-Mende (Heidelberg), Josefine Radke, Naveed Ishaque, Frank Heppner (Berlin)
ACCC: Olga Papadodima, Christos Chochos, Alex Pintzas, Georgios Zografos (Athens)
NCT-MASTER: Stefan Fröhling, Hanno Glimm (Heidelberg/Dresden)
DKFZ Proteomics Core Facility: Dominic Helm (Heidelberg)
DKFZ NGS Core Facility: Stephan Wolf (Heidelberg)
Promoter mutations: Lars Feuerbach

Pathway activities for informed decision making
Clinical decision making in molecular tumor boards, like the NCT MASTER study, mostly relies on genomic analysis (WGS/WES, RNA-seq) of patient tumors. We hypothesized that proteomic data should complement genome and transcript information, as proteins are closer to pathway activities and to drug effects.
Initially, we applied the targeted protepmics to test central kinases in pathways representative of the interventional baskets regarded within the NCT MASTER study (Wahjudi et al. 2021 Int J Cancer, 148(6):1438-51). There, proteomic data retrospectively collected from MASTER patients were used to compare recommendations based on this data, with those that had been based on genomic/transcriptomic information (WGS/WES, RNA-seq) alone. We observed that some of the original MASTER therapy recommendations were indeed supported by proteomic data. However, the protein and phosphoprotein data on pathway and drug-target activities would have suggested other treatment schemes in many other cases, some were in concordance with patient responses to the therapies having been applied to these patients. This retrospective study thus demonstrated that proteomic analysis of tumors provides information that could aid therapy decision in a molecular tumor board.
To enable the integration of high-throughput, unbiased proteomics into precision oncology, we established a mass spectrometry-based full and phospho proteome screening of tissue samples and applied it to a retrospective cohort of NCT-MASTER colorectal cancer patients, as well as patient-derived organoids. Adding the proteome to the previously acquired genomic and transcriptomic information identified new possible tumor vulnerabilities which will be validated in vitro using the patient-derived organoid models. The protein and pathway activity measurements could have a significant impact on improving the stratification of patients into more actionable treatment “baskets” and enhance personalized oncology.
Along the same lines, we have developed a computational ranking scheme (SVRACAS, available at GitHub), which aggregates and interprets multi-Omic datasets for prioritization of actionable alterations in individual patients (Vlachavas et al. 2021 Int J Mol Sci, 22(6):2822; Kontogianni et al. 2023 Cancers, 15(3):815).
Collaborations
NCT MASTER: Stefan Fröhling, Peter Horak, Hanno Glimm, Bruno Köhler (NCT Heidelberg)
MS Proteomics: Dominic Helm (DKFZ)
SVRACAS: Bruno Köhler, Peter Horak (Heidelberg), Aristotelis Chatziioannou, Konstantinos Voutetakis, Olga Papadodima (Athens), Ryangguk Kim (Washington), Rachel Karchin (Baltimore)

Reverse Phase Protein Microarrays
Molecular cancer research is driven also by advancements in technologies and tools. We have established reverse phase protein microarrays (RPPA) as a reliable and cost-effective experimental platform for quantitative protein profiling, and apply this in the tumor topics of the division and in collaborations. Furthermore, the division has been a driving force in national and international projects aimed at generating and providing tools to the scientific community.
In RPPA, samples are printed directly on solid-phase carriers. The detection of a specific protein, or a certain phosphorylation site, is carried out with a single, highly specific antibody per slide. We have adapted this approach that had been initially published by Paweletz et al. (Paweletz et al. 2001). We switched to fluorescence detection in the near infrared (NIR) range thus permitting protein profiling from as little as 20,000 cells with detection sensitivities in the fg-range (Loebke et al. 2007 Proteomics, Korf et al. 2008 Expert Opin Drug Discov). This way, less than 100 up to a few 1,000 different samples can be analyzed in parallel. Furthermore, we have advanced the RPPA technology by introducing the "RPPanalyzer" tool for data analysis (Mannsperger et al. 2010 Bioinformatics, von der Heyde et al. 2014 Biotechniques) as well with as protocols for antibody validation (Mannsperger et al. 2010 Proteome Sci, Mannsperger et al. 2011 Meth Mol Biol).
We have applied the RPPA-technology to analyze the activation status of signaling pathways, for example after RNAi-based silencing experiments (Sahin et al. 2007 PNAS), to identify protein networks regulated by miRNAs (Uhlmann et al. 2012 Mol Syst Biol), for profiling of tumor biopsy samples (Haller et al. 2008 J Pathol, Henjes et al. 2012 Oncogenesis, Wruck et al. 2015 Sci Data), to characterize drug resistance mechanisms (Borgoni et al. 2020 Cancers, Haga et al. Cancer Research 2021, Noronha et al. Cancer Discovery 2022) and to uncover protein bursts upon ligand induced cellular responses (Golan-Lavi et al. 2017 Cell Rep). Furthermore, we investigated signaling pathways associated with tumor progression and inter patient heterogeneity (Menck et al. J Exp Clin Cancer Res 2021, Menck et al. Cancers 2022).
The international ORFeome Collaboration
The long-term goal of the human genome project is to establish a comprehensive gene catalogue that contains all human genes as well as physical clones for every single gene, and the functional analysis of these genes and gene products. The International ORFeome Collaboration joins scientists from around the world who aim to generate and make widely accessible a comprehensive resource of cloned ORFs that shall cover the entire protein-coding part of the genome/transcriptome. The division Molecular Genome Analysis has been contributor to this project and has been deeply involved its further development (Wiemann et al., 2016).
The LIFEdb functional genomics resource
Based on the ORF-collection of the division Molecular Genome Analysis and the international ORFeome collaboration (see above), we generate tools for the expression of encoded proteins. These are systematically exploited to determine the subcellular localization of these proteins.
We have implemented the LIFEdb infostructure to disseminate information on the ORFeome resource (e.g., clone quality metrics) and on the utilization of this resource towards a functional annotation of encoded proteins (genes, subcellular localization data). The concept was originally described in [Simpson et al. 2000].
This project is a collaboration with the EMBL-Heidelberg (Pepperkok Team) and the UCD Dublin (Simpson Group). Data presented has been published e.g., in [Simpson et al. 2012, Laketa et al. 2007, Neubrand et al. 2005, Starkuviene et al. 2004, Simpson et al. 2001, Pepperkok et al. 2001]. LIFEdb has been described in [Mehrle et al. 2006, Bannasch et al. 2004].
The data is available here. The table provides several sort options as well as a number of links.
- GeneSymbol -> ENTREZ Gene symbol of the respective genes
- ParentCloneID -> information of the respective proteins (e.g., GO-term) - under construction
- EntryCloneID -> information on the quality and availability of clones through the ORFeome Collaboration - under construction
- NCBI -> link to sequence in EMBL/GenBank/DDBJ database
- UCSC -> link to mapping position in the UCSC genome browser
- ProteinLocalization -> experimentally determined localization of encoded proteins
- ImageFile -> link to higher resolution microscopic images of GFP-tagged fusion proteins (N- and C-terminal tagging)
(The presentation of data has been implemented by Oliver Heil)
Controls
In order to evaluate potential effects of GFP on the localization of fusion proteins we performed two types of assays.
1. We analyzed the sub-cellular localization of several fusion proteins in Vero (monkey kidney) and HeLa (human cervix carcinoma) cells (-> Cell Line Control). In every case the results obtained with the two cell lines were identical, demonstrating that the sub-cellular localization in most cases will be independent on the cell line used in the analysis. Nevertheless, we will use cell lines of different origin (e.g. hepatocytes, neuronal cells) for future experiments.
2. We analyzed the sub-cellular localization of several known proteins with predicted or known localization (-> Known Protein Control). In most cases, the localization was verified - independent of the GFP fusion part.
We conclude that the sub-cellular localization of the proteins is mostly not affected by the GFP fusion part. However, we always determine the localization of N- and C-terminal fusions (GFP-ORF and ORF-GFP since the relative orientation of the two fusion parts (ORF vs. GFP) in many cases does impact localization of either fusion-protein.
The 3of5 web application for complex and comprehensive pattern matching in protein sequences
The identification of patterns in biological sequences is a key challenge in genome analysis and in proteomics. Frequently such patterns are complex and highly variable, especially in protein sequences. They are frequently described using terms of regular expressions (RegEx) because of the user-friendly terminology. Limitations arise for queries with the increasing complexity of patterns and are accompanied by requirements for enhanced capabilities. This is especially true for patterns containing ambiguous characters and positions and/or length ambiguities.
We have implemented the 3of5 web application in order to enable complex pattern matching in protein sequences. 3of5 is named after a special use of its main feature, the novel n-of-m pattern type. This feature allows for an extensive specification of variable patterns where the individual elements may vary in their position, order, and content within a defined stretch of sequence. The number of distinct elements can be constrained by operators, and individual characters may be excluded. The n-of-m pattern type can be combined with common regular expression terms and thus also allows for a comprehensive description of complex patterns. 3of5 increases the fidelity of pattern matching and finds ALL possible solutions in protein sequences in cases of length-ambiguous patterns instead of simply reporting the longest or shortest hits. Grouping and combined search for patterns provides a hierarchical arrangement of larger patterns sets. The algorithm is implemented as internet application and freely accessible. The application is available at http://dkfz.de/mga2/3of5/3of5.html.
The 3of5 application offers an extended vocabulary for the definition of search patterns and thus allows the user to comprehensively specify and identify peptide patterns with variable elements. The n-of-m pattern type offers an improved accuracy for pattern matching in combination with the ability to find all solutions, without compromising the user friendliness of regular expression terms (Seiler et al. 2006).
Team
- Show profile

Prof. Dr. Stefan Wiemann
Head of Division
-

Sarah Burmester
-
Daniela Fischer
-
Melanie Hoos
-
Sabine Karolus
-
Dr. Cindy Körner
-
Anna MacManus
-
Christos Nousias
-
Dr. Veronica Rodrigues de Melo Costa
-
Mara Rödele
-
Luisa Schwarzmüller
-
Angelika Wörner

Selected Publications
Vlachawas E.-I. et al.
Sumer OE, Schelzig, K, et al.
Radke J. et al.
Maia, A. et al.
Wahjudi, L.W. et al.
Full List of Publications
Get in touch with us
